Informations clés pour les parents à propos de la Trofinetide (DAYBUE)
Ce document a été rédigé en collaboration avec le Rett Syndrome Research Trust (RSRT) et l’Association Française du Syndrome de Rett (AFSR). Il contient des sections rédigées par l’AFSR qui sont spécifiques à la France et qui n’étaient pas incluses dans le document original rédigé par le Rett Syndrome Research Trust.
Vous pouvez télécharger la version anglaise originale sur ce lien: Téléchargez le PDF.
This document has been written in collaboration with the Rett Syndrome Research Trust (RSRT) and the French Association for Rett Syndrome (AFSR). It contains sections written by AFSR that are specific to France that were not included in the original document authored by the Rett Syndrome Research Trust.
You can download the original English version at this link: Downlad the PDF
Par (24 mars 2023):
Monica Coenraads, Chief Executive Officer – RSRT
Randy Carpenter, MD Chief Medical Officer – RSRT
Jana von Hehn, PhD Chief Scientific Officer – RSRT
Traduction et adaptation française (26 mars 2023):
Julien Fieschi, President – AFSR
Le 12 mars 2023, la FDA (Food and Drug Administration), agence fédérale américaine chargée de la protection et de la promotion de la santé publique aux Etats-Unis, a approuvé un traitement pour le Syndrome de Rett.
Bien que le nouveau médicament DAYBUE (trofinetide/NNZ-2566) ne soit pas un remède pour le syndrome de Rett, toute annonce permettant d’envisager une amélioration fiable des symptômes invalidants du trouble est la bienvenue.
Depuis cette annonce, nous avons reçu de nombreuses demandes de parents nous demandant de les aider à comprendre les résultats de l’étude LavenderTM et nous demandant si ils auront la possibilité d’avoir accès à ce médicament en dehors des Etats-Unis.
Les familles ont du mal à comprendre les concepts statistiques et les autres informations partagées par Acadia Pharmaceuticals.
Le fond américain « Rett Syndrome Research Trust » a compilé les faits tirés des informations publiquement disponibles, y compris les résultats de l’essai LavenderTM et les informations officielles de prescription pour DAYBUE, qui sont importants à comprendre. La traduction et l’adaptation de ce document au profit des familles françaises ont été réalisé par l’Association Française du Syndrome de Rett (AFSR).
Nous espérons que les familles trouveront ce résumé utile.
RÉCAPITULATIF DES ELEMENTS CLÉS DES ESSAIS LAVENDERTM ET LILAC-1
- 61 % des patients prenant DAYBUE n’ont pas montré d’amélioration
- 13 % des patients ont été qualifiés de « nettement améliorés »
- Aucune donnée n’est fournie concernant les symptômes spécifiques qui s’améliorent
- 85 % des patients traités par DAYBUE ont eu de la diarrhée et 29 % des vomissements
- Dans l’étude où tout le monde a reçu DAYBUE, 46 % des patients se sont retirés avant la fin de l’étude
RETOUR SUR LE CALENDRIER DU DÉVELOPPEMENT DE LA TROFINETIDE
La trofinetide est le nom chimique du DAYBUE. Suite à son efficacité dans un modèle de rat pour les lésions cérébrales traumatiques (LCT), un essai clinique avec la trofinetide pour le traitement des LCT a été initié en 2008. Dans cette étude, la trofinetide a été administrée par voie intraveineuse. Elle n’a pas démontré d’efficacité pour traiter les LCT.
La trofinetide a ensuite été reformulée sous forme de solution orale et testée chez des individus souffrant de commotion cérébrale, du syndrome de Rett et du syndrome du X fragile. Les informations officielles sur la prescription du DAYBUE indiquent que son « mécanisme d’action » est inconnu. Cela signifie que la manière dont le DAYBUE produit un effet dans le syndrome de Rett n’est pas connue. Bien qu’il existe une publication rapportant l’efficacité de la trofinetide dans un modèle de souris du X fragile, il n’y a actuellement aucune publication évaluant l’efficacité de la trofinetide dans un modèle animal de Rett.
Le DAYBUE a été précédemment testé dans plusieurs essais cliniques chez des individus atteints du syndrome de Rett. Un essai clinique chez les adultes (initié en 2012) et un essai clinique en pédiatrie (initié en 2016) ont évalué sa sécurité et sa tolérabilité.
L’approbation de la FDA pour le DAYBUE est basée sur l’essai clinique LavenderTM, qui a commencé en 2019. Dans les informations de prescription, l’essai est appelé Étude 1.
| Phase | Nombre de patients | Age | Durée | Critère de jugement principal |
| 3 | 187 | 5 à 20 ans | 12 semaines | RSBQ, CGI-I |
Comme indiqué dans le tableau ci-dessus, l’essai LavenderTM comportait deux évaluations principales des résultats cliniques : RSBQ et CGI-I. Une évaluation des résultats cliniques vise à refléter comment un patient se sent, fonctionne ou survit. Le RSBQ et le CGI-I sont tous deux des échelles qui nécessitent que les aidants et les cliniciens interprètent comment leur enfant et leur patient se portent et sont donc subjectifs par nature.
QU’EST-CE QUE LE RSBQ ?
Le questionnaire comportemental sur le syndrome de Rett (RSBQ – Rett Syndrome Behavioral Questionnaire) est un sondage de 45 questions qui évalue les caractéristiques comportementales et émotionnelles du syndrome de Rett.
Voici quelques exemples de déclarations de symptômes incluses dans le RSBQ.
- Sa respiration est parfois profonde et rapide (hyperventilation).
- Des crises de cris sans raison apparente pendant la journée.
- Il y a certains jours/périodes où elle se porte beaucoup plus mal que d’habitude.
- Fait des siestes fréquentes pendant la journée.
- Visage inexpressif.
- Yeux pétillants et lumineux.
- Il y a des moments où elle est irritable sans raison apparente.
- Se berce lorsque ses mains sont empêchées de bouger.
Dans l’étude LavenderTM, le RSBQ a été utilisé pour évaluer la sévérité des symptômes de Rett à différents moments. Les aidants ont évalué chaque élément avec un score de 0, 1 ou 2 selon les critères suivants :
- 0 = l’élément n’est pas vrai pour un individu
- 1 = l’élément est parfois ou quelque peu vrai chez l’individu
- 2 = l’élément est souvent ou très vrai chez l’individu
Puisqu’il y avait 45 éléments, chacun noté de 0 à 2, le score le plus bas possible est 0 et le score le plus élevé possible est 90. Plus le score est élevé, plus la gravité des symptômes est importante.
RÉSULTATS DU RSBQ
Dans l’étude LavenderTM, 94 patients ont été assignés à recevoir un placebo et 93 patients ont été assignés à recevoir DAYBUE. Le tableau ci-dessous provient des informations de prescription de DAYBUE et présente les résultats du RSBQ.
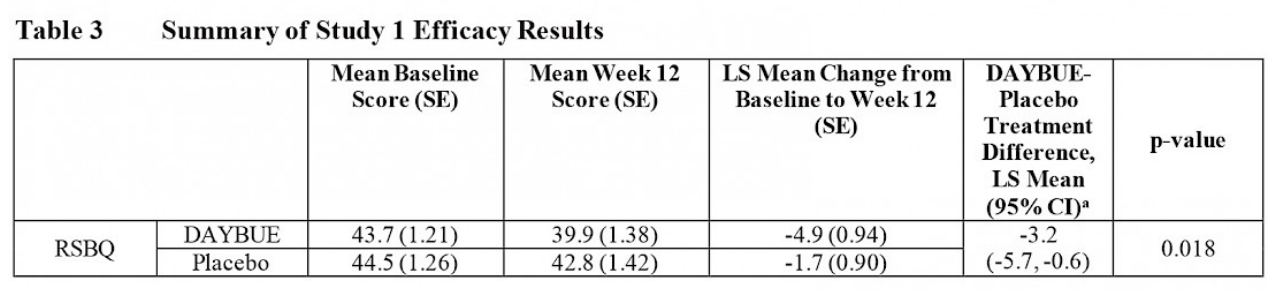
Ce tableau montre que le score moyen de base du RSBQ (au début de l’étude) était de 43,7 pour ceux prenant DAYBUE et de 44,5 pour ceux sous placebo, ce qui signifie que les scores de gravité sur le RSBQ étaient similaires pour les deux groupes avant de commencer le traitement. À la semaine 12 (qui était la fin de l’étude), le score moyen pour le groupe de patients recevant DAYBUE s’est amélioré de 4,9 points, tandis que le groupe sous placebo s’est amélioré de 1,7 point. La différence entre une amélioration de 4,9 et 1,7 point est statistiquement significative.
Ci-dessous, nous présentons les données de deux manières : une manière met l’accent sur la signification statistique de la différence entre DAYBUE et le placebo, tandis que la seconde manière se concentre sur la taille de la différence.
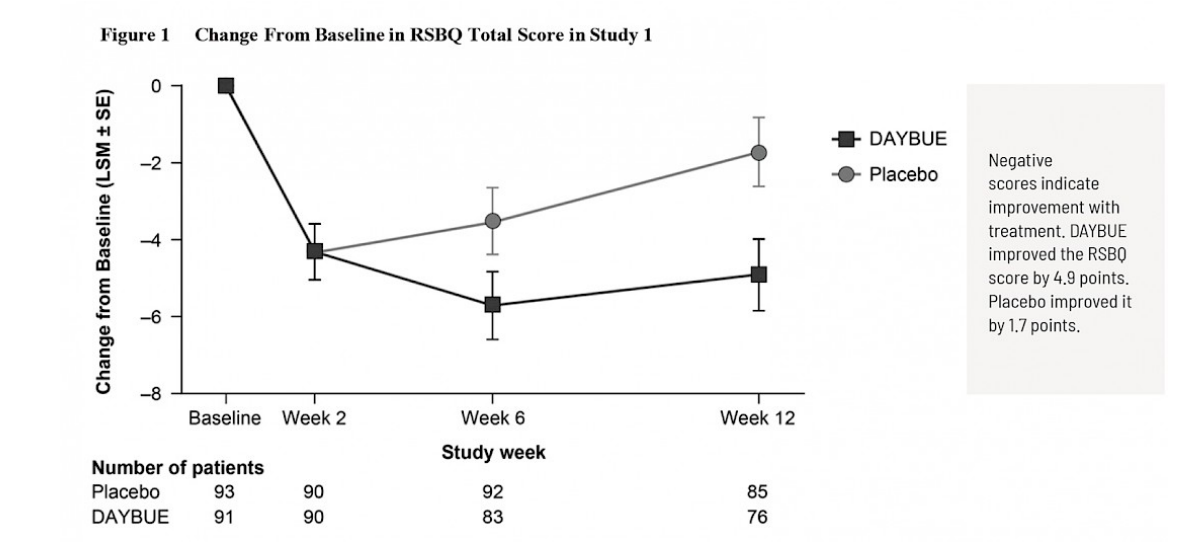
Le premier graphique est tiré des informations de prescription de DAYBUE et montre un « gros plan » du changement observé sur 8 points de l’échelle de 90 points.
Le deuxième graphique montre les mêmes données de la semaine 12, mais cette fois au lieu du changement, les scores totaux à chaque visite sont montrés sur l’échelle complète pour le RSBQ.
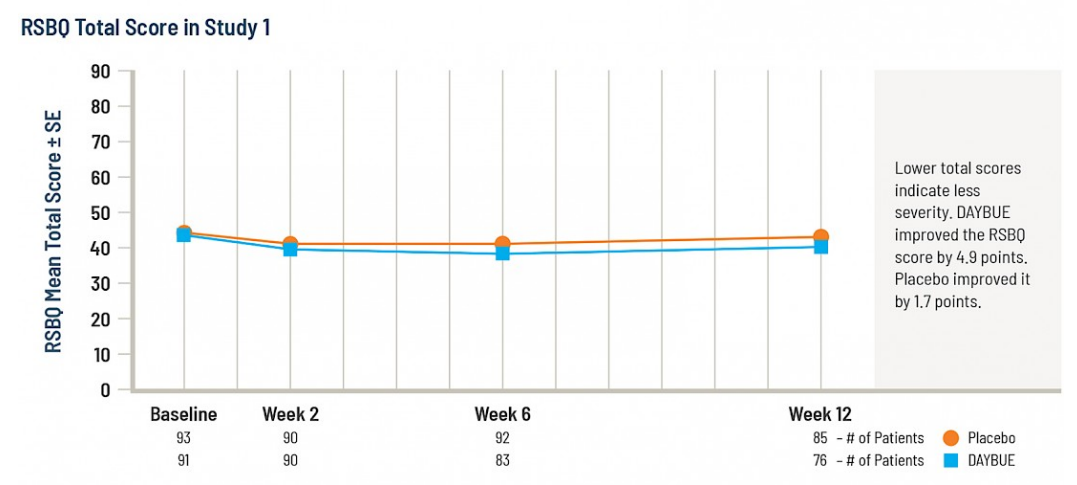
Il n’est pas possible de conclure si une différence de 3,2 points sur cette échelle est cliniquement significative, car il n’y a pas de publications qui définissent ce qui constitue un changement significatif dans le score RSBQ.
QU’EST-CE QUE LE CGI-I ?
L’échelle d’amélioration de l’impression clinique globale (CGI-I – Clinical Global Impression – Improvement) est une échelle en sept points où les cliniciens évaluent dans quelle mesure la maladie du patient s’est améliorée ou détériorée au cours des 12 semaines de traitement.
Les scores vont de 1 à 7 :
- 1 – Très nettement amélioré
- 2 – Beaucoup amélioré
- 3 – Légèrement amélioré
- 4 – Aucun changement
- 5 – Légèrement
Cliquez ici pour voir l’adaptation de l’échelle pour le syndrome de Rett qui a été utilisée dans l’étude.
RÉSULTATS CGI-I
Le tableau ci-dessous présente les résultats d’efficacité du CGI-I dans l’étude LavenderTM, extraits des informations de prescription de DAYBUE.
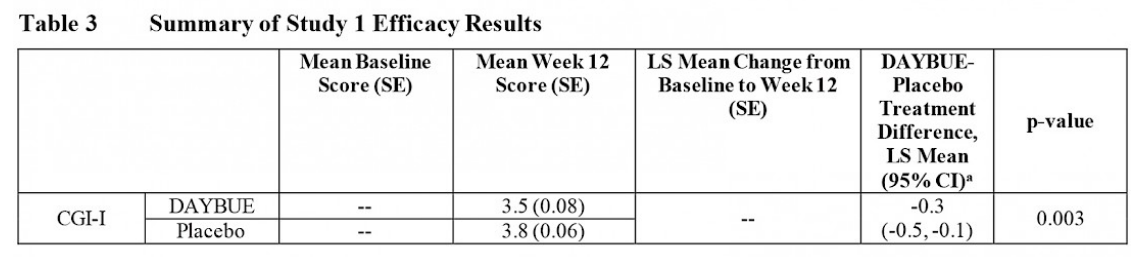
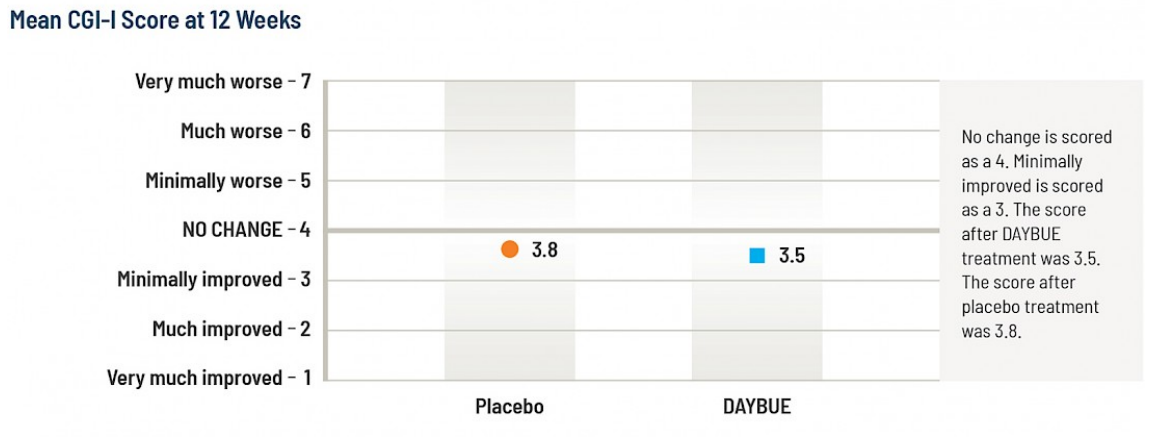
La moyenne (mean) du score CGI-I pour le groupe DAYBUE à 12 semaines est de 3,5, ce qui se situe à mi-chemin entre « aucun changement » et « légèrement amélioré », tandis que le score moyen du groupe placebo était de 3,8. La différence entre le groupe de sujets ayant reçu DAYBUE et ceux ayant reçu un placebo était statistiquement significative, en faveur de DAYBUE.
Ci-dessous, une présentation graphique de ces résultats, montrant les scores d’amélioration pour chaque groupe.
Le graphique ci-dessous tiré des informations de prescription de DAYBUE indique le pourcentage de sujets classés dans chaque catégorie spécifique du CGI-I.
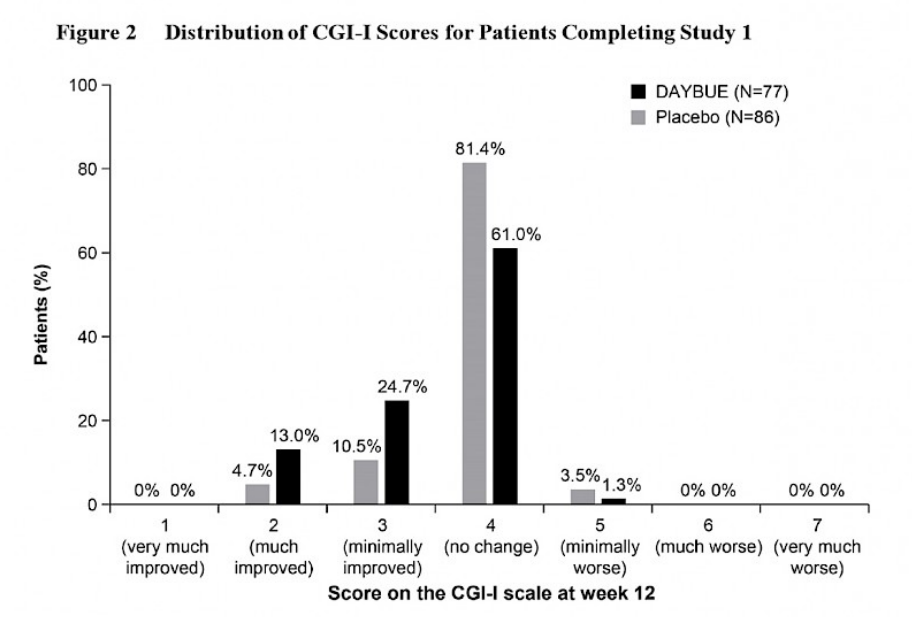
La majorité des patients, 61%, ont été classés comme « aucun changement » sous DAYBUE. Historiquement, des scores de 1 ou 2 ont été considérés comme représentant une amélioration cliniquement significative. 13% des patients sous DAYBUE et 4,7% des patients sous placebo ont été évalués comme ayant une amélioration cliniquement significative.
Ces directives expliquent ce que signifient les scores du CGI-I.
| 1 | Très nettement amélioré – presque tout va mieux ; bon niveau de fonctionnement ; symptômes minimes ; représente un changement très substantiel |
| 2 | Nettement amélioré – nettement mieux avec une réduction significative des symptômes ; augmentation du niveau de fonctionnement, mais certains symptômes persistent |
| 3 | Légèrement amélioré – légèrement mieux avec peu ou pas de réduction cliniquement significative des symptômes. Représente un très faible changement dans l’état clinique de base, le niveau de soins ou la capacité fonctionnelle |
| 4 | Aucun changement – les symptômes restent essentiellement inchangés |
| 5 | Légèrement aggravé – légèrement pire, mais peut ne pas être cliniquement significatif ; peut représenter très peu de changement dans l’état clinique de base ou la capacité fonctionnelle |
| 6 | Plus grave – augmentation cliniquement significative des symptômes et diminution du fonctionnement |
| 7 | Beaucoup plus grave – exacerbation sévère des symptômes et perte de fonctionnement |
Adapté de Spearing MK, Post RM, Leverich GS, et al. Modification de l’échelle des impressions cliniques globales (CGI) pour une utilisation dans la maladie bipolaire (BP) : le CGI-BP. Psychiatry Res 1997;73(3): 159-71.
SIGNIFICATION STATISTIQUE VS SIGNIFICATION CLINIQUE
Une différence statistiquement significative ne signifie pas nécessairement que la différence est cliniquement significative. Une amélioration cliniquement significative est une amélioration suffisante pour qu’un individu se sente mieux, fonctionne mieux ou survive mieux. De petites différences statistiquement significatives entre les groupes peuvent être des différences cliniquement significatives. Bien que DAYBUE ait obtenu une séparation statistiquement significative par rapport au placebo, ces données suggèrent que DAYBUE pourrait apporter une réduction cliniquement significative des symptômes chez un sous-ensemble limité (13%) d’individus atteints du syndrome de Rett.
QUELS SYMPTÔMES SE SONT RÉELLEMENT AMÉLIORÉS ?
Les informations de prescription indiquent que DAYBUE est indiqué pour le traitement du syndrome de Rett. Aucune information n’est fournie concernant les symptômes spécifiques qui ont été considérés comme améliorés dans l’étude LavenderTM.
EFFETS SECONDAIRES
La diarrhée et la perte de poids sont des effets secondaires importants mais potentiellement gérables de DAYBUE. La diarrhée était l’effet secondaire le plus courant, survenant chez 85% des patients prenant DAYBUE. La plupart étaient d’intensité légère à modérée. La moitié des patients avaient une diarrhée persistante ou récurrente même après des interruptions de dose, des réductions ou l’ajout de médicaments anti-diarrhéiques.
En plus de la diarrhée, des vomissements ont été signalés chez 29% des patients prenant DAYBUE. 19% des patients recevant DAYBUE ont eu des effets secondaires qui ont conduit à leur retrait de l’étude, la majorité des retraits étant dus à la diarrhée.
ARRÊTS
Les résultats du RSBQ et du CGI-I mentionnés ci-dessus sont basés sur les patients qui ont terminé l’essai. 187 patients ont commencé l’essai LavenderTM et 24 patients se sont retirés pendant les 12 semaines de traitement avant de terminer l’essai. 153 patients ont poursuivi dans l’étude en ouvert LILAC-1, où tout le monde a reçu DAYBUE pendant jusqu’à 40 semaines. Comme indiqué dans la lettre communautaire d’Acadia, 46% des patients se sont retirés de l’essai LILAC-1 avant la fin de l’étude de 40 semaines.
AUTORISATION DE MISE SUR LE MARCHE
Aux Etats-Unis, la FDA approuve les médicaments lorsque le bénéfice potentiel pour les patients l’emporte sur les risques. La FDA ne fixe pas ni ne contribue à la tarification des médicaments. L’approbation de la FDA permet aux médicaments d’être commercialisés uniquement aux États-Unis. Des approbations réglementaires distinctes sont nécessaires pour que les médicaments soient approuvés et commercialisés dans d’autres pays.
Acadia Pharmaceuticals détient les droits de commercialiser DAYBUE pour le syndrome de Rett et d’autres indications en Amérique du Nord. Selon Acadia, ils prévoient de demander l’approbation au Canada mais n’ont pas annoncé de plans pour le Mexique.
Neuren Pharmaceuticals conserve les droits de commercialisation en dehors de l’Amérique du Nord. Neuren n’a pas communiqué à ce jour sur la recherche d’approbation pour commercialiser le médicament en dehors de l’Amérique du Nord.
Dans l’hypothèse ou il le ferait, en Europe, le processus d’autorisation de mise sur le marché européen pour un médicament pour une maladie rare approuvé par la FDA (Food and Drug Administration) aux États-Unis suit généralement les mêmes étapes que pour les autres médicaments, mais il existe des procédures spéciales pour accélérer le processus pour les médicaments destinés aux maladies rares.
Les principales étapes du processus d’autorisation de mise sur le marché européen pour un médicament pour une maladie rare sont les suivantes:
- Demande d’autorisation de mise sur le marché (AMM): Le fabricant doit soumettre une demande d’AMM à l’Agence européenne des médicaments (EMA) pour obtenir l’autorisation de commercialiser le médicament dans l’Union européenne. Le dossier soumis doit inclure des données sur l’efficacité, la sécurité et la qualité du médicament, ainsi que des informations sur les maladies rares qu’il est destiné à traiter.
- Évaluation du dossier: L’EMA évalue le dossier soumis par le fabricant pour s’assurer que le médicament répond aux normes de qualité, de sécurité et d’efficacité requises. Pour les médicaments destinés aux maladies rares, l’EMA utilise une procédure spéciale appelée «procédure d’autorisation de mise sur le marché accélérée». Cette procédure permet une évaluation plus rapide des médicaments pour les maladies rares qui répondent à des besoins médicaux non satisfaits.
- Évaluation par le Comité des médicaments orphelins (COMP): Le COMP est un comité scientifique de l’EMA qui évalue les médicaments destinés aux maladies rares. Le COMP examine les données soumises par le fabricant et émet un avis sur l’autorisation de mise sur le marché. Le COMP peut également recommander que le médicament soit classé comme médicament orphelin, ce qui confère des avantages tels qu’une période de protection des données plus longue et une assistance à la recherche.
- Décision finale de la Commission européenne: La Commission européenne prend une décision finale sur l’autorisation de mise sur le marché en se basant sur l’avis du COMP. Si l’autorisation est accordée, le médicament peut être commercialisé dans tous les pays de l’Union européenne.
Le processus d’autorisation de mise sur le marché pour les médicaments destinés aux maladies rares peut être plus rapide que pour les autres médicaments en raison de la procédure d’autorisation de mise sur le marché accélérée et de la classification de médicament orphelin. Cela est dû à la reconnaissance du fait que les maladies rares nécessitent des options de traitement spéciales et que le développement de nouveaux médicaments pour ces maladies est souvent plus difficile et plus coûteux.
PRIX DU MEDICAMENT
Aux Etats-Unis, Acadia a annoncé lors de leur webcast que le prix public de DAYBUE est de 21,10 $ par mL. Cela revient à 9 495 $ par bouteille de 450 mL. Étant donné la posologie incluse dans les informations de prescription, le coût par patient, en fonction du poids, sera compris entre 385 075 $ et 924 180 $ par an. Acadia n’a pas encore annoncé s’il y aurait des réductions ou des remises sur les prix.
En France, le prix d’un médicament est réglementé et fixé par les autorités compétentes. Le processus permettant de fixer le prix d’un médicament comprend:
- L’évaluation du médicament par l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) qui analyse sa sécurité, son efficacité et sa qualité avant de l’autoriser à être commercialisé.
- Après l’autorisation de mise sur le marché, le fabricant propose un prix pour le médicament. Ce prix est basé sur différents facteurs tels que les coûts de recherche et de développement, la production et la commercialisation du médicament.
- L’ANSM évalue ensuite le service médical rendu (SMR) du médicament, c’est-à-dire son intérêt thérapeutique, pour déterminer s’il peut être remboursé par l’Assurance maladie. Le SMR est évalué en fonction de la gravité de la maladie, de la qualité des données cliniques disponibles et de l’existence ou non de traitements alternatifs.
- Si le médicament offre une amélioration par rapport aux traitements existants, l’ANSM évalue l’amélioration du service médical rendu (ASMR) pour déterminer son niveau d’amélioration. Ce niveau est utilisé pour fixer le prix du médicament.
- Le Comité économique des produits de santé (CEPS) négocie ensuite avec le fabricant pour fixer le prix final du médicament. Ce prix est basé sur le SMR, l’ASMR et d’autres facteurs tels que les coûts de production et la concurrence.
- Enfin, si le médicament est approuvé pour le remboursement par l’Assurance maladie, le remboursement est fixé par le gouvernement. Le remboursement est généralement déterminé en fonction du prix du médicament et du taux de remboursement fixé pour la classe thérapeutique à laquelle appartient le médicament.
Il est important de noter que le processus de fixation des prix des médicaments en France est complexe et implique de nombreux acteurs, notamment l’ANSM, le CEPS et les compagnies pharmaceutiques.
Le but de ce processus est de garantir l’accès à des traitements de qualité pour les patients à un coût raisonnable pour la société.
RESUME DES ELEMENTS CLÉS DES ESSAIS LAVENDERTM ET LILAC-1
- La majorité des patients, 61%, prenant DAYBUE ont été évalués comme « aucun changement » sur le CGI-I dans l’étude LavenderTM.
- 13% des patients ont été évalués comme « très améliorés » sur le CGI-I dans l’étude LavenderTM.
- Aucune donnée n’est fournie concernant les symptômes spécifiques qui s’améliorent dans l’étude LavenderTM.
- 85% des patients traités avec DAYBUE ont eu la diarrhée et 29% ont eu des vomissements. Les informations de prescription contiennent des recommandations pour gérer la diarrhée.
- Dans l’essai d’extension en ouvert, LILAC-1, 46% des patients ont arrêté l’utilisation de DAYBUE avant la fin de l’étude.
EN CONCLUSION
Il est important que chaque famille ait conscience que DAYBUE n’est pas un remède. Les données de l’essai montrent qu’il n’y aura probablement aucun changement pour la majorité des patients. Un sous-ensemble de patients peut s’améliorer. Les détails sur quels symptômes s’améliorent n’ont pas été divulgués.
En tout état de cause, cette nouvelle de l’approbation par la FDA d’un médicament pour le syndrome de Rett est bienvenue car elle augmente la visibilité du syndrome de Rett dans le monde pharmaceutique et montre qu’il existe une voie potentielle pour traiter le Syndrome de Rett. Au delà de l’effet potentiel de ce médicament sur une partie de la population, cette visibilité accrue incitera très certainement d’autres entreprises à explorer l’ajout de Rett à leurs pipelines de recherche.